Sélection Génomique
Identification de gènes de résistance pour la nodavirose chez le bar
Le séquençage de génome entier de près de 400 bars a permis la cartographie fine du phénotype de résistance à la nodavirose chez le bar européen (Dicentrachus labrax).
La combinaison de données de séquençage haut-débit avec des données de génotypages de moyenne densité (~57K marqueurs SNPs) et des données phénotypiques, a mis en évidence une association forte entre un locus de caractère quantitatif (QTL) du groupe de liaison 12 (LG12) et la survie des bars face à une infection à la nodavirose (Figure 1, Delpuech et al. 2022).
A partir de l'annotation du génome de bar, disponible en libre accès sur les bases de données publiques, nous avons pu identifier deux gènes d'intérets dont ZDHHC14 et IFI6. La mise en évidence d'une protéine induite par un interféron dans les mécanismes de survie à la nodavirose est une avancée considérable dans le domaine.
Les écloseries peuvent dès lors utiliser ces connaissances pour l'amélioration de la sélection génomique de leurs lignées dans le but de développer des bars plus résistants.
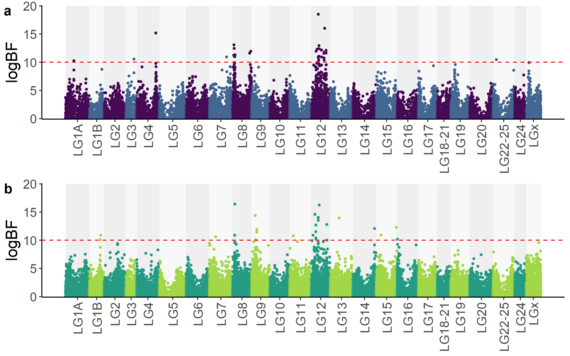
Figure 1 extraite de Delpuech et al. (2022), bioRxiv.
Manhattan plot représentant les associations génétiques-phénotypes obtenues à l'issu de l'étude de la résistance à la nodavirose chez le bar européen.
Etudes des valeurs génétiques pour la sélection de caractère d'intérêts
La sélection génomique a été utilisée pour améliorer de très nombreux caractères d'intérêts possédant une hératibilité assez élevée. Par exemple, ce type d'approche a été appliqué avec succès à de nombreuses espèces d'élevages, et récemment sur des espèces aquacoles pour la résistance à des pathogènes. Elle a permis un gain génétique de 12,5 % en moyenne par génération pour la résistance à différentes maladies. La sélection génomique est donc une approche prometteuse pour lutter contre des maladies affectant les populations sauvages comme les populations domestiques (Griot et al. 2021).
D'autre part, dans un article de référence pour la prédiction génomique, Aliakbari et al. 2021 ont mis en évidence une prédiction génomique similaire en utilisant soit (i) un jeu de données dit d'entraînement comprenant des animaux de lignées génétiquement apparentées, soit (ii) un ensemble d'entraînement provenant de la population ciblée. De plus, dans les cas où il est possible de combiner des données apparentées et des données de la population cible, la précision ne pourra qu'augmenter pour des caractères (Figure 3).
Ces résultats de génétique quantitative donnent une meilleure connaissance pour la conception des populations de référence, en particulier pour initier la sélection génomique dans des populations où le nombre d'échantillons historiques est faible.
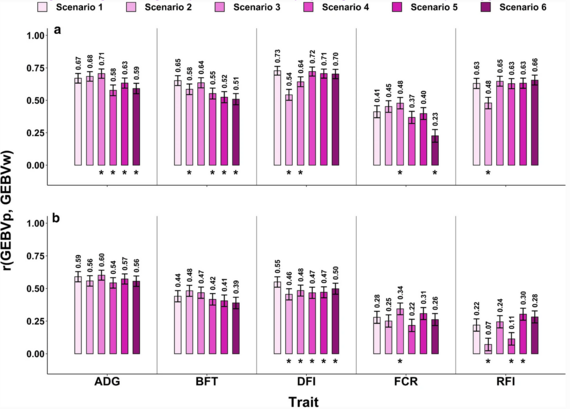
Figure 3 extraite de Aliakbari et al. (2021).
Corrélations entre GEBVp et GEBVw, et leurs barres d'erreur pour les lignes HRFI (a) et LRFI (b).
*=Différence significative avec le scénario 1 (contrôle) selon le test t de Williams à un niveau de 0,05.
RFI=apport alimentaire résiduel, ADG=gain moyen journalier, FCR=indice de consommation, DFI=apport alimentaire journalier, BFT=épaisseur du gras dorsal.
Où nous trouver ?
LDgenX SAS
SIREN 952193837 - RCS Montpellier - NAF 7112B